


北京时间10月9日17时45分,2024年诺贝尔化学奖揭晓。
DeepMind 创始人 Demis Hassabis 和高级成员 John Jumper,以及华盛顿大学教授 David Baker,三位学者凭借蛋白质预测系统 AlphaFold 和蛋白质结构预测荣获今年的化学奖。
人工智能为何能屡获国际医学大奖?很多生物学家都有些不屑,认为这里没有先进的科学,只有工程学。
事实上,AlphaFold的发展之路并不顺利。从积累已确认的蛋白质结构数据到预测几乎所有人类蛋白质的结构,AlphaFold 为蛋白质结构研究提供了新的视角。

获奖者介绍:DeepMind 创始人 Demis Hassabis 和高级会员 John Jumper、华盛顿大学教授 David Baker。
获奖理由:表彰他们对蛋白质结构设计和预测的贡献。

1. 一个难题
众所周知,蛋白质在细胞内发挥着许多重要作用,其功能与其三维结构形态密切相关。对蛋白质结构的深入了解可以揭示其功能并解决生物学之谜。

克里斯蒂安·安芬森
60多年前,美国国立卫生研究院(NIH)已故科学家克里斯蒂安·安芬森(Christian Anfinsen)发现了一个有趣的现象:蛋白质无需外界帮助即可自行恢复特定形状。
基于此,他得出结论:蛋白质的形状是由其成分氨基酸的顺序决定的。
事实上,即使是很小的蛋白质,尝试所有可能的形状也可能比宇宙存在的时间还要长。但在生物细胞内,蛋白质可以在几毫秒内快速找到最合适的形状。
这让我们很困惑,大自然是如何解决这个问题的呢?
为了了解蛋白质是如何构建并探索其结构,研究人员使用了多种方法。他们试图用能量方程来表达物理相互作用,同时使用 X 射线晶体学和其他技术来创建可以作为蛋白质结构蓝图的模板。此外,他们结合了有关特定氨基酸(例如它们是否带电荷)及其在链中位置的知识,以获得有关蛋白质结构特征的线索。

1994年,马里兰大学的John Moult和他的团队启动了结构预测临床评估(CASP)项目,以跟踪蛋白质结构预测领域的进展。该项目每两年组织一次,参与者收到实验室已解决但尚未发表的蛋白质氨基酸序列,然后应用自己开发的系统生成预测模型,然后与实验结果进行比较并评分。
在过去的几十年里,模型性能以小幅增量缓慢上升,有时停滞甚至倒退。

华盛顿大学教授 David Baker 早期开发了一种方法,从全球共享的蛋白质数据库 (PDB) 中提取短片段来预测蛋白质内的局部结构。虽然肯定是有益的,但这种基于片段的策略不仅耗时,而且对绝大多数蛋白质的应用也有限。
与此同时,科学家们一直在积累已证实的蛋白质结构的数据。截至2014年,PDB中已有超过10万条蛋白质结构记录,但这些众多的蛋白质结构记录只是当时已知的数千万条蛋白质序列中的一小部分。
2、将人工智能引入折叠过程
在2018年第13届CASP竞赛中,DeepMind团队利用机器学习技术改进了预测方案。与依赖预设逻辑的传统人工智能方法不同,机器学习可以自我发现数据中的模式。在使用机器学习作为蛋白质预测网络的核心组件后,Hassabis 和 Jaber 的团队在高精度上遥遥领先,甚至比去年的最佳水平提高了近 50%。
Science 报道 DeepMind 在 CASP13 上的表现
尽管取得了成功,DeepMind 研究人员并没有停下脚步:他们希望开发出误差不大于原子大小的实用工具。哈萨比斯、贾比尔和其他人集思广益,将几何和遗传学概念融入到已知的蛋白质知识中,例如具有特定半径的原子和具有特定角度的键。他们的目标是在不干扰系统自主学习能力的情况下考虑这些因素。
研究人员设计了从有限的实验证据中提取最多信息的方法,并采用了使 AlphaFold2 能够高效学习的策略。它们允许 AlphaFold2 在任何阶段进行调整,以避免早期错误。在整个过程中,可以逐步细化结构模型。
Hassabis、Jaber 和同事还放弃了指导传统算法的原则,例如线性邻近性,而是更多地关注三维关系,因为相隔数百个亚基的氨基酸可以在折叠蛋白质中共存。此外,他们开发了一种算法,特别关注不同氨基酸的物理距离。
AlphaFold2所取得的突破并不是单一要素的贡献,而是众多创新理念共同作用的结果。
3、严格培训
为了训练该系统,Hassabis 和 Jaber 的团队使用蛋白质数据库 (PDB) 对已识别的结构进行实验。 AlphaFold2反复将其模拟答案与真实答案进行比较,最终成为现实。对训练集中的每个成员重复此过程后,算法吸收了蛋白质结构的原理。
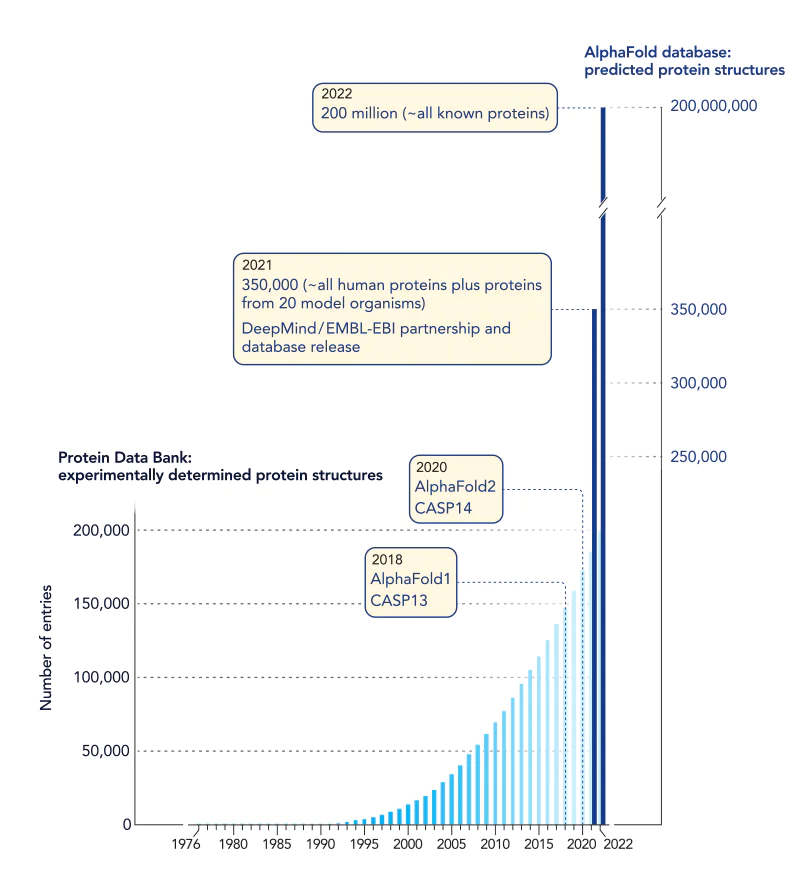
过去几年,机器学习将蛋白质结构领域推向了一个新领域
研究人员使用了一些技巧来提高网络的学习能力。例如,他们将氨基酸隐藏在 MSA 中,并要求它填补空白。这样,他们就需要人工智能系统掌握进化关系的规则。它们还递归地提供任何给定步骤的输出,这为 AlphaFold2 提供了许多重新考虑和改进的机会。
AlphaFold2 还计算其预测的置信度,这些置信度分数使研究人员能够从可用数据中提取更多信息,从而提高其性能。
研究人员向 AI 系统输入大约 140,000 个 PDB 序列后,它会运行另一组结构未解析的序列。此外,他们选择了最可靠的 350,000 个序列/结构对,并根据这些数据训练系统,从而提高其准确性。
4. 重塑蛋白质科学
2020年,AlphaFold2在CASP14竞赛中脱颖而出。它的预测达到原子精度,并且可以在几分钟内为没有模板的蛋白质生成出色的结果,这使其成为在不知道类似结构的情况下构建高分辨率预测的第一种方法。
2021 年 7 月,Hassabis 和 Jaber 发布了他们对几乎所有人类蛋白质的模型和结构预测。短短两年时间,他们论文的影响力就超过了《自然》自 1900 年以来发表的近 10 万篇研究论文的几乎全部,并且这篇论文在顶级期刊中排名第 50 位。被超过 7,000 篇论文引用。

哈萨比斯和贾比尔随后与欧洲分子生物学实验室的欧洲生物信息学研究所合作,与科学界共享该程序和数据库,超过一百万研究人员已经使用了这些资源。
此后,DeepMind 团队将其目录扩展到基因组已测序的生物体中几乎所有已知的蛋白质。这些目录包括流行病威胁病毒和世界卫生组织高度优先病原体的蛋白质组。
目前,AlphaFold2系统已经在多个生物医学领域及相关领域产生了巨大影响。通过应用 AlphaFold2,研究人员重新设计了针对人类细胞的蛋白质,并为药物输送和基因治疗开辟了新途径。学术实验室和公司正在致力于利用 AlphaFold2 开发疫苗、设计药物和制造分解污染物的酶,未来是光明的。
参考:
AlphaFold—用于预测蛋白质结构。拉斯克基金会。